
Quick links
- Overview
- The condition
- Treatment
- Current research in RPE65-retinal degeneration
- Practical advice
- Referral to a specialist centre
- Further information and support
- A patient’s perspective
- References
- RPE65 gene card: for professionals
Overview
Mutations in the RPE65 gene lead to progressive degeneration of the retinal photoreceptor (light-sensing) cells in children. It is one of the major causes of Leber congenital amaurosis and a small proportion of retinitis pigmentosa cases.[1] Night blindness or difficulty adapting to dark conditions from early childhood is a prominent symptom. Other features include involuntary movement of the eyes (known as nystagmus), reduced central visual sharpness (visual acuity) and loss of peripheral vision of variable severity. Despite difficulties seeing in the dark, children may still retain some vision in bright environments up to early teenage years before progressive deterioration thereafter but this can vary among patients within and between families. As of 2019, UK patients with retinal degeneration due to mutations in RPE65 can be treated with a form of gene therapy called Luxturna under the National Health Service (NHS).
The condition
Symptoms
The condition usually starts during early childhood, sometimes even as young as within the first few months of life, where parents may notice poor visual behaviour in their child which is usually accompanied by nystagmus. Night blindness is a prominent symptom in which the child may seem afraid of being in the dark or dim environment but tends to behave normally during daytime or in a brightly-lit room. Furthermore, there is normally a reduction in visual sharpness and loss of peripheral vision but some may have sufficient vision to function normally in a well-lit environment during their infant and primary school years.[2-5]
Vision tends to remain stable up to early teenage years before deteriorating progressively thereafter, leading to “tunnel vision” which is eventually lost by middle age.[3] The severity of these symptoms and the rate of visual decline are highly variable, but children with earlier onset of visual symptoms tend to be more severely affected.[6] Rarely, some patients may only start experiencing symptoms from adolescence onwards in which case visual function might not deteriorate as rapidly as usual.[7-9]


Cause
The RPE65 gene instructs cells to produce a protein known as retinoid isomerase. It helps the photoreceptor cells in the retina to convert incoming light into electrical signals that are transmitted to the brain to generate sight through a series of chemical process called the visual cycle. When the RPE65 gene is not working properly, this process cannot be undertaken which leads to sight loss.

How is it diagnosed?
The condition is diagnosed based on the symptoms, family history and clinical evaluation of the retina. As it can appear similar to inherited retinal degenerations caused by other genes, the diagnosis can only be confirmed with genetic testing by identifying mutations in the RPE65 gene.
How is it inherited?
1) Autosomal recessive (AR) inheritance
Most cases of RPE65-related retinal degeneration are inherited in this manner. Both parents are usually unaffected carriers (who only carry one faulty copy of the gene) while the patient has two faulty gene copies (inherited one each from each parent). This means that every newborn has the following risks regardless of gender:
- 25% chance of being affected by the condition
- 25% chance of being unaffected and not a carrier
- 50% chance of being a carrier with no symptoms

2) Autosomal dominant (AD) inheritance
A very small number of cases are inherited in this manner. Only one copy of the faulty gene (inherited from either parent) is required to cause disease. This means that each newborn of the patient has a 50% chance of inheriting the condition regardless of gender. It usually causes a later-onset disease with less severe visual deterioration than if there are two faulty copies of the RPE65 gene.
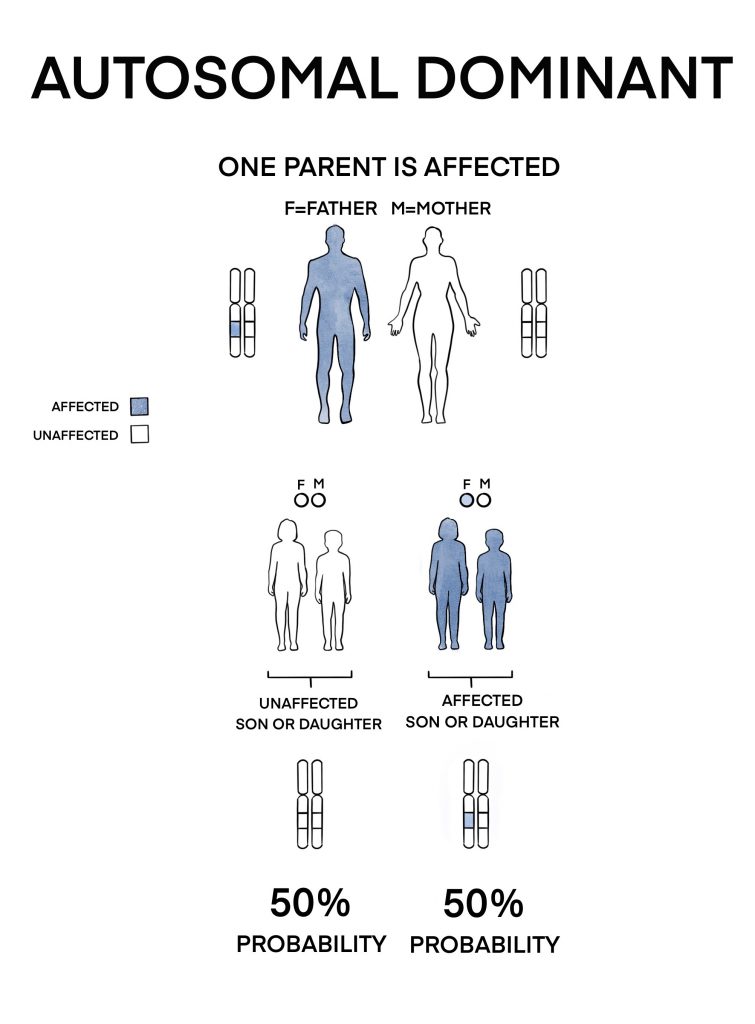
If you or your child is affected by RPE65-related retinal degeneration, it is advisable to see a genetic counsellor to obtain more information and advice on inheritance and family planning options.
Related links
Treatment
Is there any treatment?
1) Gene therapy
Gene therapy aims to halt retinal degeneration by replacing the mutated gene with a normal healthy copy. This enables the affected cells to regain some of their function and produce functioning proteins.
Patients with RPE65-related retinal degeneration are now able to receive a treatment called Luxturna (voretigene neparvovec) under the NHS. In Luxturna, a normal copy of the RPE65 gene is “packaged” into a harmless virus called the adeno associated virus (AAV) which is then surgically injected into the retina (sub-retinal injection). This way, the affected retinal cells are maximally exposed to the viruses containing the normal gene.

This was introduced following the success of a clinical trial where patients treated with Luxturna in both eyes (sequentially at different periods) have improved visual function and navigational abilities at low light levels compared to those who did not receive treatment.[10] This improvement has been sustained up to 4 years after the initial surgery.
In the UK, patients can be referred to the following treatment centres for consideration of Luxturna treatment:
- Great Ormond Street Hospital for Children, London (for children < 10 years of age)
- Moorfields Eye Hospital, London (for children and adults)
- Manchester Royal Eye Hospital
- Oxford Eye Hospital
The main goal of Luxturna is to halt progressive retinal degeneration and thus stabilises remaining vision. The positive results reported from the aforementioned trial may not be observed in every patient and some may not be eligible for treatment as there may be insufficient healthy photoreceptor cells remaining or other factors. Therefore, we strongly emphasise that interested patients or parents should have a thorough discussion with an ophthalmologist familiar with this procedure before proceeding.
Related links
- Safety of gene therapy
- Approval of Luxturna by the UK National Institute of Clinical Excellence (NICE)
- NHS news on the first UK patients receiving Luxturna treatment
- Explanation of different phases of clinical trials
2) Supportive treatment
For those who may not want to proceed with gene therapy or not eligible, treatment is focused on limiting additional damage to the retina and optimising remaining sight:
- Regular eye examinations to correct any long- (hyperopia) or short-sightedness (myopia)
- Referral to low vision services
- Visual aids and assistive technology
- Have a healthy diet consisting of fresh fruits and green leafy vegetables
- Wear UV protected sunglasses in bright light
- Using blue light screen protectors on mobile devices or computer screens*
*Current available evidence shows that blue light emitted from screens do not damage the retina but it can disrupt our sleep cycle. The screen protectors are used as a precautionary measure.
3) Optimising development
Visual impairment can have a negative impact on a child’s early general development. Therefore, timely referral to practitioners familiar with developmental surveillance and intervention for children with visual impairment (VI), such as developmental paediatricians as well as a Qualified Teacher of children and young people with Visual Impairment (QTVI) is crucial to optimise their developmental potential.
The Developmental Journal for babies and young children with visual impairment (DJVI), developed by Great Ormond Street Hospital Developmental Vision team is a structured early intervention programme designed to track developmental and vision progress in children from birth to three years of age. It is mainly used by qualified healthcare professionals working in services providing support to babies and young children with VI in conjunction with the child’s parents. Children with VI may be referred to developmental services such as the developmental vision clinic in the Great Ormond Street Hospital for Children or other specialist developmental services for further management.
Related links
Current research in RPE65-retinal degeneration
1) Gene therapy
Although Luxturna is an approved gene therapy for patients, there are several other ongoing clinical trials to improve the safety and efficacy of treatment:
- NCT05858983: This study is a multi-centre, open-label, phase I/II clinical study to evaluate the safety, tolerability, efficacy, immunogenicity, and in vivo biodistribution characteristics of FT-001 in subjects with biallelic RPE65 mutation-associated retinal dystrophy. Assessments will include visual acuity, vector shedding, immunogenicity and adverse events. Participants will be monitored for 5 years after treatment.
- NCT04516369: This is an open-label, single-arm study to evaluate the safety and efficacy of bilateral subretinal administration of AAV2-hRPE65v2 in Japanese patients with biallelic RPE65 mutation-associated retinal dystrophy. Assessments will include full-field light sensitivity threshold testing, visual fields, visual acuity, vector shedding, immunogenicity and adverse events. Participants will be monitored for 5 years after treatment.
- NCT00999609: The study is a Phase 3, open-label, randomized controlled trial of gene therapy intervention by subretinal administration of AAV2-hRPE65v2. At least twenty-four subjects, three years of age or older, will be recruited. The intervention group will receive AAV2-hRPE65v2 at either The Children’s Hospital of Philadelphia or University of Iowa to determine if it improves visual and retinal function in individuals with RPE65 gene mutations.
- NCT00481546: A Phase I clinical trial to evaluate the safety of subretinal administration of a rAAV2-CBSB-hRPE65 in individuals with RPE65-associated retinal disease. This open-label trial includes five cohorts with different age groups. Ocular and systemic toxicity will be closely monitored before and after vector administration to assess potential adverse effects. The study aims to determine the safety of this gene transfer approach and its implications for treating various retinal degenerative diseases.
- NCT03602820: Multi-site, non-randomized, observational study, for up to 15 years after subretinal AAV2-hRPE65v2 administration for each subject. The study is a non-interventional, follow-up study of subjects who participated in previous AAV2-hRPE65v2 gene therapy clinical trials.
Safety of gene therapy
The safety of gene therapy, particularly utilising voretigene neparvovec (VN), which is delivered through subretinal injection, is a topic of ongoing investigation and does come with potential risks and complications.
Short term:
The most commonly reported short term side effects were limited to the injected eye and related to the injection procedure itself. These include inflammation, transiently elevated eye pressure and retinal tears, all of which resolved either with eye drops or laser treatment. Serial blood tests showed minimal immune response from the body, but some cases of vitritis and outer retinal infiltrates are managed with immunosuppressive therapy.
Intermediate term:
More recent studies of the real-world experience of patients have shown there is a risk of progressive chorioretinal atrophy, although functional outcomes such as visual acuity and visual fields have been suggested to remain stable. Atrophic retinal changes begin at the injection site and can start to occur 2 weeks after the injection. While the atrophy may affect areas within and outside the subretinal bleb. The presence of paracentral scotomas related to atrophy in a subset of cases indicates potential localised functional impacts. Regular assessments and imaging techniques, such as fundus autofluorescence, play a crucial role in monitoring these intermediate-term risks, allowing for early detection and management strategies to mitigate adverse effects and optimise the safety profile of gene therapy.
Long term:
The long-term safety profile of gene therapy necessitates extended scrutiny and understanding. Over a follow-up period spanning several years, concerns have been raised about the persistence and development of chorioretinal atrophy. However, functional outcomes, including visual acuity and visual fields, remain stable at 6-8 years despite the observed atrophy. This paradox underscores the complexity of the long-term effects, suggesting that while anatomical changes may manifest, the functional benefits of the treatment appear to endure. Continued, comprehensive long-term monitoring is required to gain deeper insights into the safety and efficacy of gene therapy and for informing potential refinements in treatment approaches over extended periods.
Related links
- Explanation of different phases of clinical trials
- Research Opportunities at Moorfields Eye Hospital UK
- Searching for current clinical research or trials
Practical advice
Living with RPE65-retinal degeneration
Patients are still able to lead fairly independent lives through maximising their available vision and having access to social support. Here are some ideas:
- Attending the low vision clinic which provides access to low vision specialists, Eye Clinic Liaison Officers (ECLOs), visual aids and visual rehabilitation services
- Utilising assistive technologies that can improve quality of life
- Getting in touch with the local education authority for access to qualified teachers for children with visual impairment (QTVI) and special educational needs co-ordinator (SENCO)
- Registering your child as sight impaired (SI) or severely sight impaired (SSI) if eligible for access to social support and financial concessions
- Getting in touch with national or local charities for advice and peer support
Related links
Referral to a specialist centre
If you are based in the UK and would like to be seen in the nearest specialist centre for your eye condition, either to receive a more comprehensive genetic management or just to find out more about current research, you can approach your GP to make a referral or alternatively arrange for a private appointment.
More information can be found in our “How to see a genetic eye specialist?” page.
Further information and support
- Retina UK
- Royal National Institute of Blind People (RNIB)
- Guide Dogs for the Blind Association
- Look UK
- VICTA
A patient’s perspective
A family’s experience with RPE65 associated LCA and how treatment with Luxturna changed their lives
References
- Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci U S A. 1998;95(6):3088-3093
- Thompson DA, Gyürüs Pt, Fleischer LL, et al. Genetics and Phenotypes of RPE65 Mutations in Inherited Retinal Degeneration. Investigative Ophthalmology & Visual Science. 2000;41(13):4293-4299
- Chung DC, Bertelsen M, Lorenz B, et al. The Natural History of Inherited Retinal Dystrophy Due to Biallelic Mutations in the RPE65 Gene. Am J Ophthalmol. 2019;199:58-70
- Weleber RG, Michaelides M, Trzupek KM, Stover NB, Stone EM. The phenotype of Severe Early Childhood Onset Retinal Dystrophy (SECORD) from mutation of RPE65 and differentiation from Leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2011;52(1):292-302
- Li S, Xiao X, Yi Z, Sun W, Wang P, Zhang Q. RPE65 mutation frequency and phenotypic variation according to exome sequencing in a tertiary centre for genetic eye diseases in China. Acta Ophthalmol. 2019
- Zahid S, Branham K, Schlegel D, et al. Retinal Dystrophy Gene Atlas. Springer; 2018
- Bowne SJ, Humphries MM, Sullivan LS, et al. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet. 2011;19(10):1074-1081
- Hull S, Holder GE, Robson AG, et al. Preserved visual function in retinal dystrophy due to hypomorphic RPE65 mutations. Br J Ophthalmol. 2016;100(11):1499-1505
- Jauregui R, Park KS, Tsang SH. Two-year progression analysis of RPE65 autosomal dominant retinitis pigmentosa. Ophthalmic Genet. 2018;39(4):544-549
- Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849-860
Updated on January 31, 2024