
Quick links
- Overview
- The condition
- Treatment
- Current research in aniridia
- Practical advice
- Referral to a specialist centre
- Further information and support
- A patient’s perspective
- References
- Aniridia: for professionals
Overview
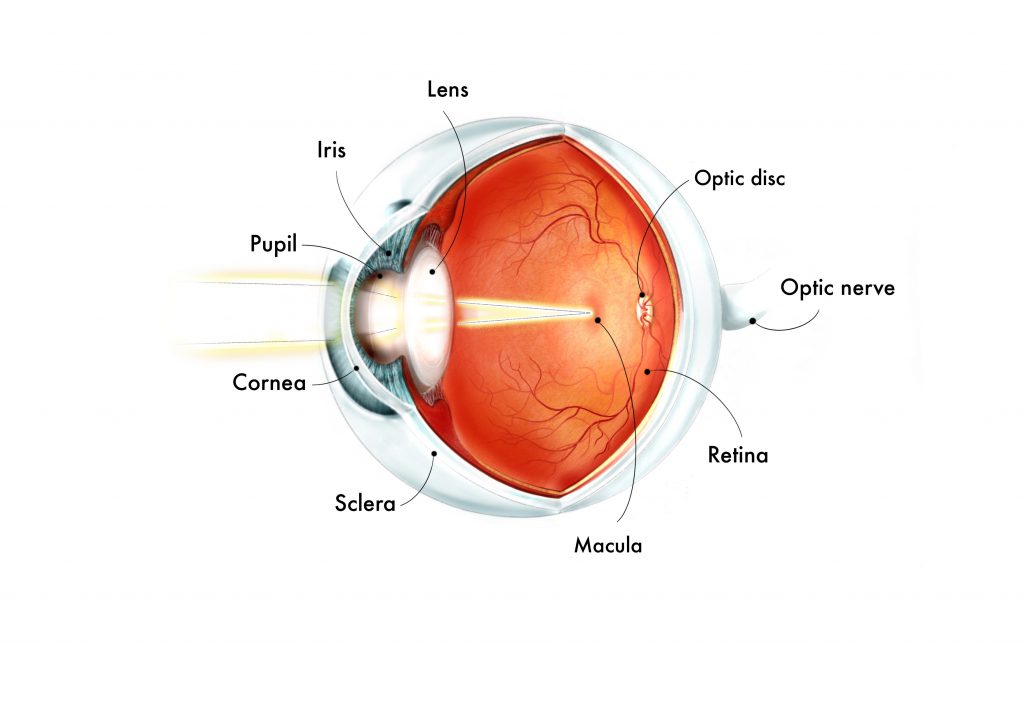
Aniridia is a rare birth eye defect affecting 1 in 40,000 to 100,000[1] people. It is caused by genetic changes to the PAX6 gene, resulting in but not limited to an under-developed or absent iris. Other structures of the eye could be affected as well, such as cornea, the trabecular meshwork, lens, fovea, optic nerve and overall size of the eye (microphthalmia).
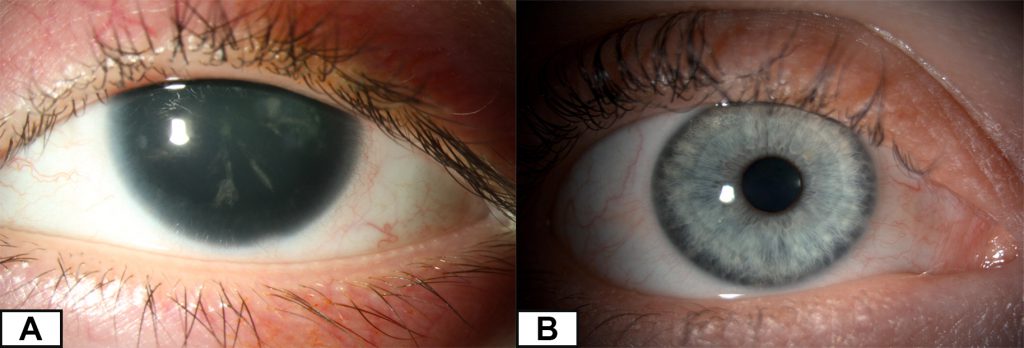
Patients experience a variety of symptoms such as glare, severe light sensitivity, poor vision and involuntary movement of the eyes (also known as nystagmus). Aniridia can either be inherited from a parent or develop spontaneously in an individual (when developing in the womb during pregnancy) without prior family history. The condition varies in severity among patients, even within the same family but individuals usually show little variability between the two eyes.
Aniridia is typically an isolated eye condition. However, in one-third[2] of cases it is associated with a condition that affects other parts of the body as well. This condition is known as the Wilms tumor, aniridia, genitourinary anomalies and range of developmental delay (WAGR) syndrome due to mutations involving both PAX6 and WT1 genes. Affected individuals are at increased risk of developing Wilms tumour, a form of childhood kidney cancer.[3]
The condition
Symptoms
1) Glare or light sensitivity
A normal iris can control the amount of light entering into the eye by changing the size of the pupil (central black hole of the eye), reducing glare and sensitivity to light in bright conditions. Patients with aniridia are not able to do this as the iris tissue is not formed properly. As a result, patients experience severe light sensitivity which sometimes causes headaches.

2) Poor vision from a young age
- Incomplete development of the fovea
Medically, this is known as foveal hypoplasia. The fovea is responsible for central and colour vision and is usually identified by a “central dip” on a specialised scan called optical coherence tomography (OCT). This “central dip” is absent in fovea hypoplasia.

- Incomplete development of the optic nerve
Medically, this is known as optic nerve hypoplasia. The optic nerve is a structure behind the eye that relays electrical signals to the brain to generate images. With an under-developed optic nerve, the visual processing part of our brain is not able to receive information from our retina, leading to sight loss.
3) Progressive visual deterioration
- Cataract
- Glaucoma
- Aniridia-related keratopathy (ARK)
The PAX6 mutation associated with aniridia disrupts the function of stem cells in the cornea.[4][5] As a result, the stem cells are not able to regenerate and replace old or damaged cells, leading to dryness and difficulty recovering from minor injuries to the eye. Eventually, the cornea becomes scarred and opaque (not clear) from the migration of non-transparent conjunctival tissue and its blood vessels.

4) Other systemic issues
- Affected sense of smell
- Hearing difficulties
- Behavioural problems
- Sleep disorders
- Obesity
- Type 1 diabetes (rarely)
Cause
In aniridia that is only localised to the eyes (isolated aniridia), 99%[2] of cases are caused by mutations in the PAX6 gene. The gene provides instructions for the development of multiple vital organs during early pregnancy including the eyes and the brain.[1] The ELP4 gene has been implicated in very rare cases.[2]
How is it diagnosed?
Aniridia is usually discovered at birth or during infancy. The eye doctor is able to diagnose this by examining your child. Genetic testing is performed to exclude WAGR syndrome and to look for genetic changes in the PAX6 and ELP4 genes.
How is it inherited?
1) Autosomal dominant inheritance
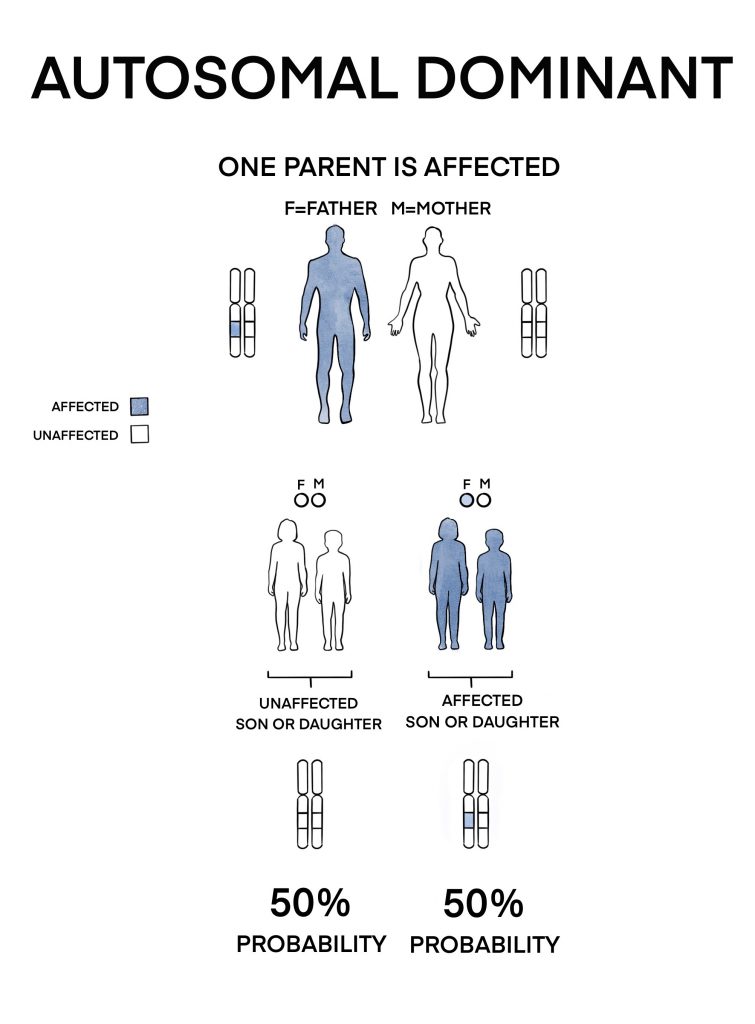
Two-thirds[2] of isolated aniridia cases are inherited from a parent through this pattern. Each newborn of an affected individual has a 50% chance on having the condition.
2) Sporadic (occurring spontaneously)
This occurs in one-third of isolated aniridia cases. Due to the variability of aniridia, it can be very mild in some people and not cause any symptoms. Therefore, parents of a patient with presumed sporadic aniridia should undergo genetic testing to confirm that they do not carry the faulty gene. Individuals with sporadic aniridia may pass on to their children in an autosomal dominant pattern.
If you or your child is affected by aniridia, it is advisable to see a genetic counsellor to obtain more information and advice on inheritance and family planning options.
Related links
Treatment
Is there any treatment?
There is currently no treatment for aniridia but researches are exploring various approaches, one of which has entered clinical trial. In the meantime, here are some ways to help alleviate symptoms and treating eye conditions associated with aniridia:
1) Visual development
- Regular eye examinations to correct short (myopia) or long-sightedness (hyperopia) with glasses
- Patching therapy to avoid development of a “lazy eye”
2) Glare or light sensitivity
- Wearing hats when out in bright daylight
- Wearing tinted or photochromic glasses
- Wearing tinted, coloured or artificial pupil contact lenses (might not be suitable for every patient due to aniridia-related keratopathy)
- Placing sunlight diffusers at back windows of a car
3) Cataract
- Cataract surgery can be undertaken if severely impacting vision
- Surgery can be more complicated due to aniridia
- Vision improvement might be limited due to other associated eye conditions

4) Glaucoma
- Regular check-up with a glaucoma specialist to ensure eye pressure is well controlled and therefore preserve sight
- Eye drops are usually first line of treatment
- Surgery may be considered if not responding to eye drops
5) Aniridia-related keratopathy
- Frequent use of eye drops to keep corneal surface moist
- Corneal transplant surgery may be considered if scarring affects the centre of the cornea, causing severe impact to vision. A careful and thorough discussion with your ophthalmologist is required to see if it is beneficial.
Children newly diagnosed with aniridia are often assumed to potentially have WAGR syndrome until proven otherwise by genetic testing. As it can also be associated with hearing difficulties, behavioural issues, sleep disorders, obesity and sometimes type 1 diabetes, they will be referred to a geneticist and a paediatrician for further assessment and monitoring.
Current research in aniridia
1) Nonsense suppression therapy
Nonsense suppression therapy is a new drug-based treatment targeting conditions caused by nonsense mutations. A nonsense mutation introduces an abnormal “stop” signal into a gene that halts protein production prematurely, resulting in a protein which is too short and not functional. A drug called ataluren (TranslarnaTM) modifies the affected cell to “ignore” these abnormal “stop” signals and produce normal full-length functional protein.
Up to 40%[6] of patients with aniridia are due to nonsense mutations. Animal studies have shown that ataluren eye drops is able to reverse the developmental defects associated with aniridia when given at a specific time frame after birth.[7][8]
In a pre-clinical study led by Professor Moosajee, researchers created eye structures and cell models using induced pluripotent stem cells in the lab to mimic reduced PAX6 protein levels. The drug Amlexanox showed promise in helping cells produce more of the full-length PAX6 protein. This drug worked well in both the 3D eye structures and the 2D cell models, correcting the abnormalities associated with the protein deficiency. These findings suggest that cells derived from induced pluripotent stem cells could be useful for studying this condition, and that Amlexanox might be a potential treatment aniridia.
A clinical trial called Study of Ataluren in Participants With Nonsense Mutation Aniridia (STAR) was conducted to assess the safety and effectiveness of oral ataluren in patients with aniridia due to nonsense mutations, however, the trial failed to meet its primary endpoints.
Related links
2) Limbal stem cell transplant
Limbal stem cells
In a normal healthy cornea, the cells on the superficial layer called the epithelium are maintained by a group of stem cells located in the limbus. These cells reside in a highly specialised structure called the limbal niche. It provides an environment for the stem cells to survive and function optimally. During normal shedding of the epithelium or when injured (such as a scratch in the eye), the stem cells multiply to replace the shed or damaged cells. They also keep the cornea transparent by preventing migration of the non-transparent conjunctival tissue.


In aniridia patients, mutation in the PAX6 gene disrupts the limbal niche and subsequently affects the function of the stem cells.[4]This leads to ARK and when it is severely impacting vision, limbal stem cell transplant surgery may be required.
Types of transplant
- Allografts
Allografts are tissues harvested from either a living-related relative or a cadaver. As anirdia usually affects both eyes, conventional transplant surgeries for ARK require stem cells to be harvested from these sources before embedding it on a patient’s eye.[9]The long-term outcomes of these surgeries are variable.[10][11] In addition, patients with allografts require long-term medications to suppress the body’s immune system so that the transplanted cells are not rejected.
- Cultivated oral mucosal epithelial transplantation (COMET)
To avoid using such potent medications, COMET was developed where cells lining the patient’s own oral cavity are harvested and cultured in the laboratory to transform into corneal epithelium. The transformed cells are subsequently transplanted onto the patient’s eye. The visual outcome of this technique was not optimal as the transplanted epithelium was thicker and less transparent compared to normal corneal epithelium.[12]
Scientists believe that the variable results seen with conventional limbal stem cell transplant surgeries are partly due to their failure to restore the highly specialised limbal niche. Therefore, RAFT, a bioengineered mould made from a type of natural human protein called collagen typically found in skin was developed to mimic the limbal niche.[13]A clinical trial for ARK patients, funded by the Medical Research Council (MRC) is due to start in 2020.
3) Anti-inflammatory and tear film treatment
When transplanted cells or tissues are rejected by the body, it’s often due to inflammation. Studies are exploring drugs that target specific immune cells to manage this inflammation, such as CTLA4-Ig, which blocks certain receptors on immune cells. For eye-related inflammation, using artificial tears with anti-inflammatory properties can be a first step. Also, drops made from a person’s own blood, called autologous serum eye drops, have been successful in treating inflammation and abnormal blood vessel growth in the eye. Starting treatment early with these drops could help slow down the progression of certain eye conditions, but it requires careful selection of patients and long-term monitoring because the conditions usually worsen slowly over time.
An ongoing phase 1 clinical trial (NCT05400590) is investigating the safety and efficacy of growth factor-enriched plasma (GFP) and autologous serum (AS) in patients with aniridia.
4) Anti-angiogenic treatment
A potential treatment for aniridia-associated keratopathy (AAK) involves using medications that prevent abnormal blood vessel growth, these are called anti-VEGF medications. Examples include bevacizumab, which is applied directly to the eye, and ranibizumab and aflibercept, however the latter two are not officially approved for this specific use. Another drug called GS-101 (Aganirsen) is being tested in clinical trials. It targets a specific protein involved in blood vessel growth (insulin receptor substrate-1). These medications not only slow down the progression of AAK but may also help transplanted cells survive better after surgery. Studies are also looking into different treatments such as photodynamic therapy and UVA-based corneal collagen crosslinking. The main goal is to create a healthy area around the transplanted cells where they can survive and function well for a long time.
5) Artificial iris prosthesis
These are devices that are surgically inserted into the eye to alleviate glare and to improve cosmetic appearance. The three main types of prosthesis are:
- CustomFlex Artificial Iris (HumanOptics AG)
- Morcher Iris diaphragm (Morcher® GmbH)
- 311 Aniridia Lens II (Ophtec)
The CustomFlex Artificial Iris was recently approved for clinical use in the USA.
Although these devices might improve quality of life, we do not advocate them due to the potential sight threatening complications that might arise. These can include:
- Development or progression of glaucoma (most common)[14]
- Development or worsening of ARK
- Prolonged inflammation in the eye which might lead to glaucoma
- Retinal detachment
- Severe eye infections
- Repeated surgeries if the device is out of position
Related links
- Research Opportunities at Moorfields Eye Hospital UK
- Searching for current clinical research or trials
Practical advice
Living with aniridia
Patients are still able to lead fairly independent lives through maximising their available vision and having access to social support. Here are some ideas:
- Attending the low vision clinic which provides access to low vision specialists, Eye Clinic Liaison Officers (ECLOs), visual aids and visual rehabilitation services
- Utilising assistive technologies that can improve quality of life
- Noticing your child’s preferential head position when looking at objects can help you to understand where you should hold toys or position yourself as this is often where your child finds to have the best vision or least glare
- Contacting the local council’s social services department for access to rehabilitation services and assist with practical home adaptations
- Getting in touch with the local education authority for access to qualified teachers for children with visual impairment (QTVI)
- Registering as sight impaired (SI) or severely sight impaired (SSI) if eligible for access to social support and financial concessions
- Getting in touch with national or local charities for advice and peer support
Related links
- Coping with sight loss
- Education and learning
- Employment support
- Family support service
- Driving and alternative transport
Referral to a specialist centre
If you are based in the UK and would like to be seen in the nearest specialist centre for your eye condition, either to receive a more comprehensive genetic management or just to find out more about current research, you can approach your GP to make a referral or alternatively arrange for a private appointment.
More information can be found in our “How to see a genetic eye specialist?” page.
Further information and support
- Aniridia Network
- International WAGR Syndrome Association (IWSA)
- Royal National Institute of Blind People (RNIB)
- Guide Dogs for the Blind Association
- Look UK
A patient’s perspective
References
- Hingorani M, Hanson I, Van Heyningen V. Aniridia. European Journal of Human Genetics. 2012;20(10):1011
- Moosajee M, Hingorani M, Moore AT. PAX6-Related Aniridia. In: GeneReviews®[Internet]. University of Washington, Seattle; 2018.
- Breslow NE, Norris R, Norkool PA, et al. Characteristics and outcomes of children with the Wilms tumor-Aniridia syndrome: a report from the National Wilms Tumor Study Group. J Clin Oncol. 2003;21(24):4579-4585.
- Shortt AJ, Secker GA, Munro PM, Khaw PT, Tuft SJ, Daniels JT. Characterization of the limbal epithelial stem cell niche: novel imaging techniques permit in vivo observation and targeted biopsy of limbal epithelial stem cells. Stem Cells. 2007;25(6):1402-1409.
- Ramaesh K, Ramaesh T, Dutton GN, Dhillon B. Evolving concepts on the pathogenic mechanisms of aniridia related keratopathy. Int J Biochem Cell Biol. 2005;37(3):547-557
- Lee H, Khan R, O’Keefe M. Aniridia: current pathology and management. Acta Ophthalmol. 2008;86(7):708-715.
- Gregory-Evans CY, Wang X, Wasan KM, Zhao J, Metcalfe AL, Gregory-Evans K. Postnatal manipulation of Pax6 dosage reverses congenital tissue malformation defects. J Clin Invest. 2014;124(1):111-116.
- Wang X, Gregory-Evans K, Wasan KM, Sivak O, Shan X, Gregory-Evans CY. Efficacy of Postnatal In Vivo Nonsense Suppression Therapy in a Pax6 Mouse Model of Aniridia. Mol Ther Nucleic Acids. 2017;7:417-428
- Fernandez-Buenaga R, Aiello F, Zaher SS, Grixti A, Ahmad S. Twenty years of limbal epithelial therapy: an update on managing limbal stem cell deficiency. BMJ Open Ophthalmol. 2018;3(1):e000164.
- Baylis O, Figueiredo F, Henein C, Lako M, Ahmad S. 13 years of cultured limbal epithelial cell therapy: a review of the outcomes. J Cell Biochem. 2011;112(4):993-1002.
- Shortt AJ, Secker GA, Notara MD, et al. Transplantation of ex vivo cultured limbal epithelial stem cells: a review of techniques and clinical results. Surv Ophthalmol. 2007;52(5):483-502.
- Yazdanpanah G, Haq Z, Kang K, Jabbehdari S, Rosenblatt ML, Djalilian AR. Strategies for reconstructing the limbal stem cell niche. Ocul Surf. 2019;17(2):230-240
- Levis HJ, Kureshi AK, Massie I, Morgan L, Vernon AJ, Daniels JT. Tissue Engineering the Cornea: The Evolution of RAFT. J Funct Biomater. 2015;6(1):50-65
- Srinivasan S, Ting DS, Snyder ME, Prasad S, Koch HR. Prosthetic iris devices. Can J Ophthalmol. 2014;49(1):6-17